Computational description of a Diels-Alder reaction
March 2026 update — tl;dr set up a computational chemistry workflow, wrote a tool to fragment products into starting geometries, found a transition state and verified it.
Status: proof of concept. I'd like to benchmark more reactions from Loco et al.; transition states for reactions B2 and B3 were found in the same way as below, but I haven't looked into calculating pre-complex energies yet. Along the way I wrote a script which separates a Diels-Alder product along the direction of bond formation into a plausible starting point for a relaxed surface scan. It will not work well for reactions where the transition state is not very similar to the product and does not guarantee endo approach, but is less annoying to use than trying to position starting materials by hand.
The reaction between acrylonitrile and cyclopentadiene (reaction B1 in Loco et al., 2022) was selected as a sort of proof-of-concept that my setup was actually capable of running the relevant computations. One of the combinations benchmarked and recommended by the paper is the ωB97XD functional with a 6-31G basis set because it’s cheap and accurate. I used the ωB97X with D3BJ dispersion instead (yes, they are different…) with the same basis set because ORCA doesn’t provide the ωB97XD functional.
A relaxed surface scan was conducted first in order to find an initial structure close enough to the transition state. (This took 13 hours to complete because I turned off my laptop at some point before it finished, but I expect it would have taken around 2 hours undisturbed.) I had Claude plot the energy against the scanned coordinate to identify the structure most similar to the transition state.
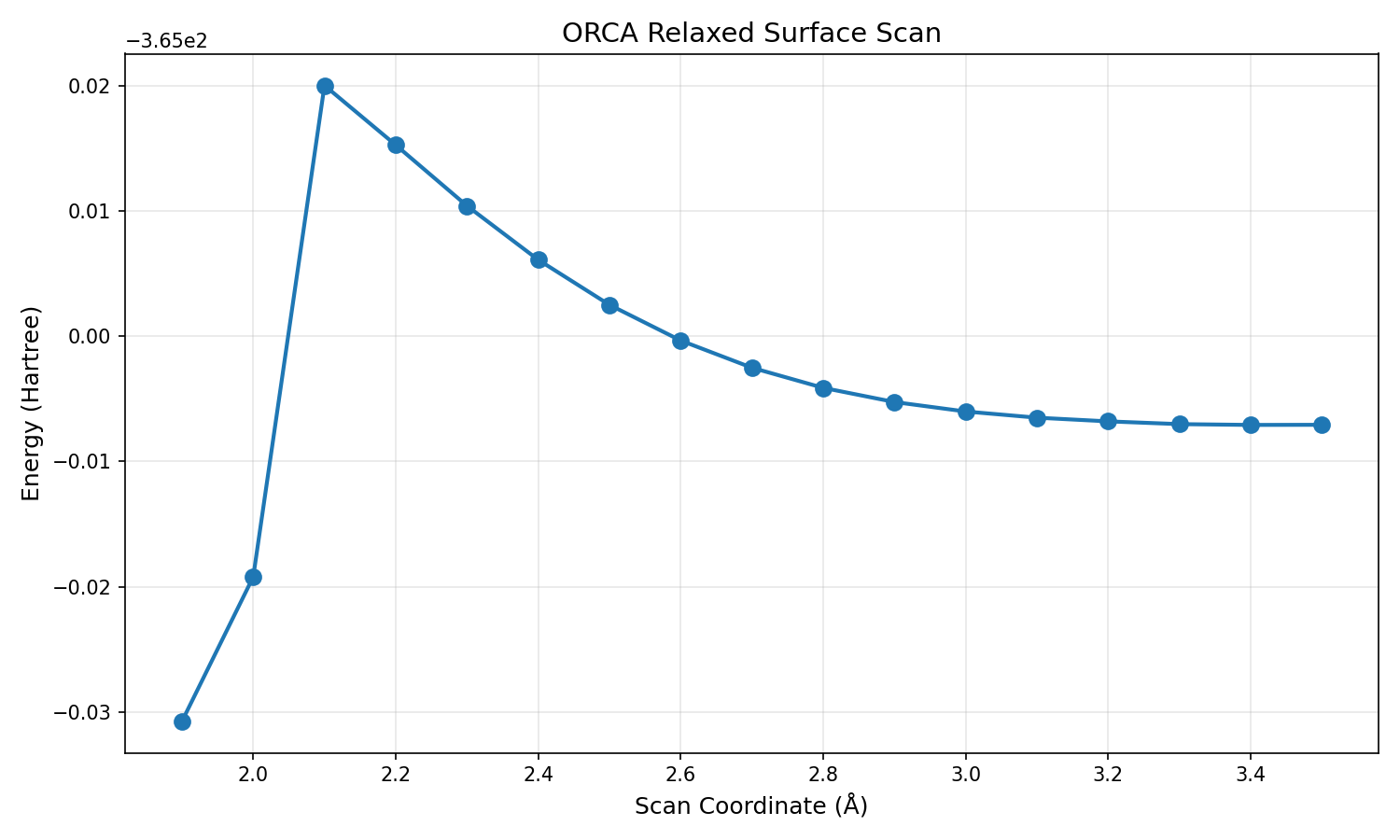
The Avogadro file corresponding to the maximum energy configuration (2.1A along the scan coordinate) was used for a transition state optimisation, which completed in around 13 minutes. Both of my previous transition state searches hit the maximum number of optimisation cycles after an hour, so I was hopeful that the program had produced something interesting this time. In order to verify a transition state, one conducts a frequency calculation in ORCA; transition states should have exactly one imaginary vibrational frequency. This was not immediately obvious to me, but here is my understanding of how it works: on a potential energy surface (or PES, a plot of energy against $ 3N-6 $ nuclear coordinates), transition states are saddle points lying between two local minima. Analogous to how local minima are critical points with no negative second derivatives, transition states are critical points with exactly one negative second derivative. The connection between this and the frequency analysis is that chemical bonds, like springs, have specific ways in which they can vibrate, and they do so at specific frequencies
\[v \propto \sqrt{\frac{k}{m}}.\]Just as with classical springs, $ k $ is the second derivative of energy with respect to (nuclear) position, and therefore the vibrational frequency for exactly one vibrational mode will be the square root of a negative number – an imaginary frequency. The results of the frequency calculation are shown below (note that ORCA uses the negative sign (-) for imaginary numbers).
| Mode | Frequency (cm⁻¹) | Note |
|---|---|---|
| 4 | 0.00 | |
| 5 | 0.00 | |
| 6 | -543.33 | imaginary mode |
| 7 | 93.21 | |
| 8 | 136.37 | |
| 9 | 260.99 | |
| 10 | 279.50 |
So that’s definitely the transition state. It’s kind of exciting to have a representation of this thing in front of me because they dictate a lot of synthetic chemistry. We’ll still need to find pre-complex energies and ideally re-run the energy calculations on all four basis sets before drawing comparisons to the paper’s benchmarks though. Maybe a project for another time.
Failure log
B1 - acrylonitrile with cyclopentadiene
Transition state optimisation run 1: Maximum cycles reached, result was product-like
Relaxed surface scan run 1: Unclear what went wrong but energy at 1.8A should not be higher than the transition state. Also energy barrier is much larger than reported by Loco et. al.
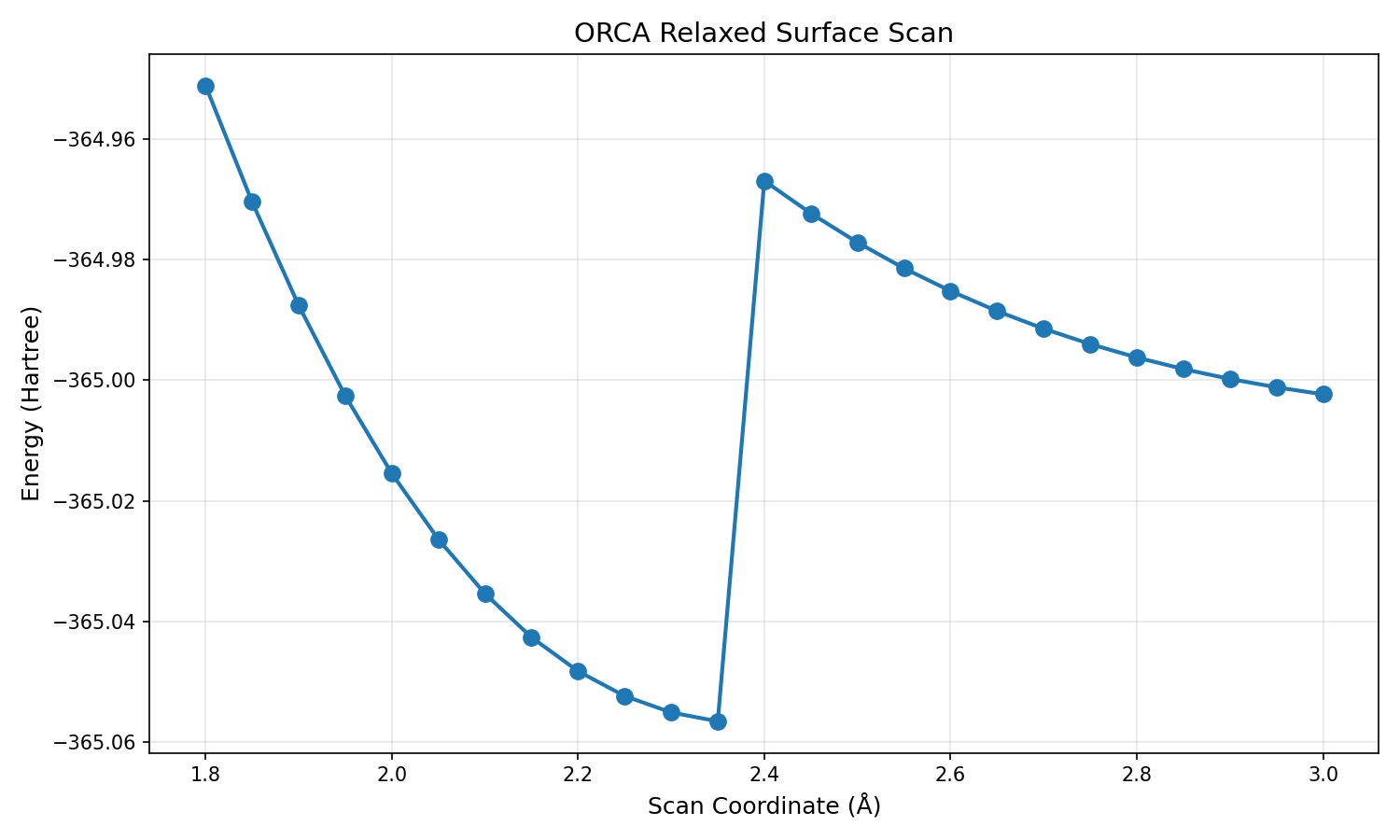
Transition state optimisation run 2 - Didn’t use the result from the relaxed surface scan, but initial structure was poorly drawn. Maximum cycles reached, terminated at an aziridine.
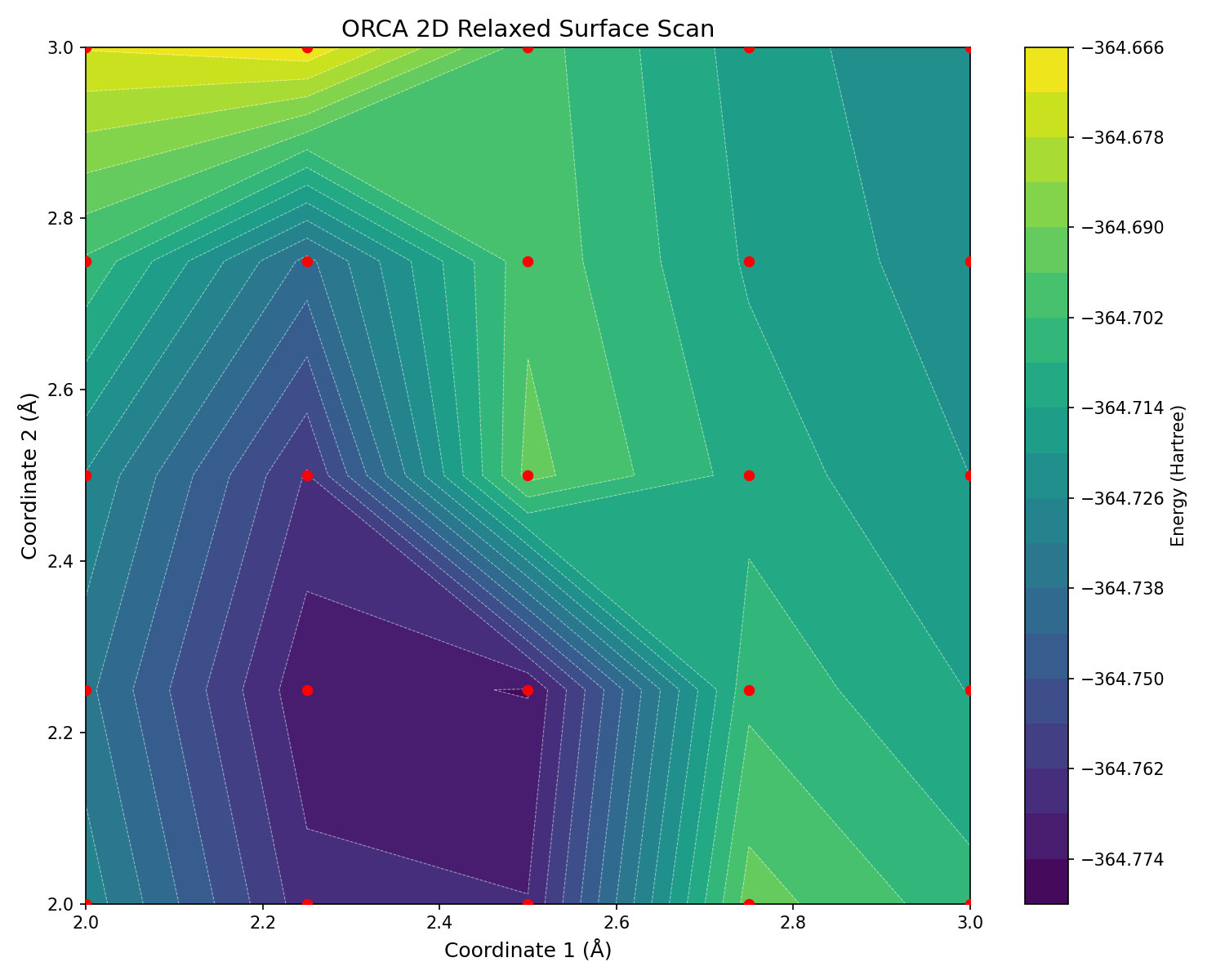
Relaxed surface scan run 2 (2D): Same as previous surface scan, energy barrier is more reasonable but still much larger than expected.
References
- Loco, D., Chataigner, I., Piquemal, J.-P., & Spezia, R. (2022). Efficient and Accurate Description of Diels-Alder Reactions Using Density Functional Theory. ChemPhysChem, 23(18), e202200349. https://doi.org/10.1002/cphc.202200349